
Abstract
Angiomatoid fibrous histiocytoma (AFH) is an exceptional, soft tissue neoplasm of indeterminate lineage and intermediate malignancy associated with minimal localized tumour reoccurrence and infrequent distant metastasis. Preliminarily contemplated to be a variant or derivative of malignant fibrous histiocytoma or undifferentiated pleomorphic sarcoma or an unusual fibrohistiocytic sarcoma, angiomatoid fibrous histiocytoma predominantly incriminates young po pulation and superficial sites although several extra-somatic sites can be implicated.
Author Contributions
Academic Editor: Qiping Dong, China.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2021 Anubha Bajaj
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Preface
Angiomatoid fibrous histiocytoma (AFH) is an exceptional, soft tissue neoplasm of indeterminate lineage and intermediate malignancy associated with minimal localized tumour reoccurrence and infrequent distant metastasis. The neoplasm is preliminarily contemplated to be a variant or derivative of malignant fibrous histiocytoma or undifferentiated pleomorphic sarcoma or as an unusual fibro-histiocytic sarcoma. A neoplasm of young population, angiomatoid fibrous histiocytoma predominantly emerges within superficial sites although several extra-somatic sites can be implicated. Angiomatoid fibrous histiocytoma is a translocation- associated neoplasm demonstrating repetitive chromosomal rearrangements with EWSR1-CREB1, EWSR1-ATF1 and FUS –ATF1 genetic fusions. The neoplasm can be adequately determined with cogent morphology, ancillary cytogenetic and molecular genetic analysis.
Disease Characteristics
Of ambiguous lineage, the neoplasm was originally scripted by Enzinger as a malignant sarcoma and designated as angiomatoid “malignant fibrous histiocytoma”1. However, the neoplasm demonstrates an intermediate biological behaviour, is associated with minimalistic localized tumour reoccurrence of around 15% and distant metastasis in roughly 1% to 5% subjects. The exceptional neoplasm configures nearly 0.3% of soft tissue neoplasia2. Constituent elliptical or spindle -shaped cells are of obscure aetiology and genesis and may not phenotypically simulate a specific subtype of mature mesenchymal cell.
Tumefaction is commonly encountered within the superficial dermis or subcutaneous tissue of extremities. Lesions within head and neck are frequent2. However, neoplasms within non-somatic sites as the skeletal muscle, bone, orbit, vulva, brain, omentum, retroperitoneum, ovary, mediastinum and pulmonary parenchyma are infrequent2.
Majority of instances appear within 30 years although no age of disease incrimination is exempt and lesions appear in infants or up to ninth decade. Children and young adults are commonly implicated and congenital lesions are encountered. Somatic neoplasms commonly emerge at 12 years to 18 years. Enlarged extra-somatic neoplasms are associated with mean age of disease representation at 35 years2. A specific gender predilection is absent wherein the myxoid variant demonstrates a slight female predominance2, 3. Molecular genetic analysis of angiomatoid fibrous histiocytoma characteristically displays genomic rearrangements of EWSR1 gene in a majority of neoplasms, indicative of EWSR1-CREB1 genetic fusion occurring due to t(12;22)(q33;q12) chromosomal translocation 3, 4.
EWSR1-ATF1 genomic rearrangements appearing due to chromosomal translocation t(12;22)(q13;q12) or FUS-ATF1 genetic rearrangements arising due to chromosomal translocation t(12;16)(q13;p11) are infrequent. Cogent molecular analysis for EWSR1 chromosomal rearrangements is beneficial in neoplasms with atypical clinical or histological manifestations3, 4. Angiomatoid fibrous histiocytoma with focal myxoid alteration is documented in around 14% instances. Significant deposition of myxoid substance wherein an estimated 60% of neoplasm is composed of myxoid areas is exceptional and constitutes below <5% of angiomatoid fibrous histiocytomas.
Myxoid variant appears in extremities of young adults wherein nearly 50% neoplasms occur within the upper extremity. Myxoid angiomatoid fibrous histiocytoma is minimally aggressive and approximately 14% neoplasms are associated with localized tumour reoccurrence whereas proportionate distant metastasis is unknown3, 4.
Disease Pathogenesis
Chromosomal translocations are preliminary alterations engendering the neoplasm with consequent genetic fusions which frequently configure contemporary, tumour- specific, chimeric transcription factors thereby initiating genetic dysregulation. Propensity of pertinent partner genes in concurrence with malignant metamorphosis is ambiguous although may appear on account of proximity of three dimensional nuclear chromosomal arrangements. Engendered genetic fusion products commences diverse manifestations in concordance with specific, unexplained target cells generating the neoplasm in combination with secondary genetic and epigenetic events which contribute to neoplastic conversion.
As circumscribing lymphoid cellular proliferation is immune reactive to desmin, a myoid or myofibroblastic cellular lineage is postulated which aids designation of the neoplasm as angiomatoid myosarcoma4, 5.
Fibroblastic reticulum cells contribute to a structural, supporting framework within lymph node stroma wherein a subset of cells are immune reactive to desmin. Pleuripotent mesenchymal stem cells are posited to be potential cells generating several subtypes of sarcoma. Lesions previously classified as sclerosing haemangioma, dermatofibroma or fibrous histiocytoma are now reclassified as angiomatoid fibrous histiocytoma3, 4.
Clinical Elucidation
The exceptional tumefaction recapitulates adjunctive vascular, fibrohistiocytic and myofibroblastic neoplasms. Classically, systemic symptoms as anaemia, weight loss, malaise, chills and pyrexia are demonstrated which may subside following tumour resection. Clinical symptoms are cytokine-mediated, a phenomenon which is frequently observed in extra-somatic neoplasms4, 5.
Clinical symptoms can be site-specific and the indolent, gradually progressive, nodular tumefaction is seldom painful or tender. Lesions can be discovered incidentally or clinically misinterpreted as a haematoma or haemangioma4, 5.
Angiomatoid fibrous histiocytoma is occasionally documented to concur with adjunctive malignancies such as disseminated testicular carcinoma, Hodgkin’s lymphoma, retroperitoneal neuroblastoma, vulval metastasis of endometrial and ovarian carcinoma or nasopharyngeal carcinoma. In aforesaid instances, multiple tumour reoccurrences predominantly to regional lymph nodes and exceptionally to pulmonary parenchyma, liver and brain are observed. Although associated with extended survival and complete alleviation, tumour associated morbidity due to distant metastasis can appear following two decades of initial disease representation4, 5.
Genetic confirmation of primary angiomatoid fibrous histiocytoma with accompanying metastatic expression is rare.
Localized tumour reoccurrence is contingent to infiltrative tumour perimeter or inadequately resected tumefaction located within the head and neck. Extra-somatic neoplasms demonstrate an enhanced, proportionate localized reoccurrence, in contrast to somatic neoplasms. Tumour invasion to deep-seated fascia or skeletal muscle is concurrent with localized and distant tumour metastasis4, 5.
Histological Elucidation
On gross examination, a firm, multinodular, multi-cystic, haemorrhagic, well circumscribed, encapsulated tumefaction is discerned. Tumour diameter varies from 2 centimetres to 4 centimetres although magnitude extends up to 10 centimetres with a median diameter of 2.5 centimetres. Cut surface is solid or partially cystic, greyish/yellow, whitish or reddish with thickened cyst walls impacted with haemorrhagic fluid4, 5.
Characteristically, the multinodular neoplasm is composed of elliptical histiocytoid, spindle-shaped or spherical cells with abundant, eosinophilic cytoplasm. Proliferating cells configure a tumour syncytium composed of sheets, whorls and fascicles. Centrically disseminated pseudo-angiomatous vascular spaces are layered with neoplastic cells and appear devoid of a true endothelial layer. Foci of stromal haemorrhage or hemosiderin pigment deposits are encompassed within a thick, fibrous, pseudo-capsule along with variable peri-capsular lymphocytic and plasma cell infiltrate5, 6. Antecedent lesions of angiomatoid fibrous histiocytoma exemplify diverse stages of vascular articulations layered with endothelium, basal lamina and occasional pericytes5, 6.
The circumscribed, lobulated or multinodular neoplasm is encompassed within a dense, incomplete, fibrous pseudo-capsule. Majority (80%) of neoplasms are encompassed with an intense, peripheral lymphocytic and plasma cell infiltrate. Lymphoid cuff may exhibit germinal centres.
Predominantly multinodular, solid areas display a reticular arrangement. Intersecting strands and cords of tumour cells are intermixed within a variably myxoid to collagenous stroma5, 6.
Cystic, reticulated areas configuring around 20% of the neoplasm are comprised of sheets of uniform, histiocyte- like cells with a peripheral storiform or swirling tumour pattern. Cystic tumour zones delineate enlarged, distended, vascular spaces coated by singular to multiple layers of inconspicuous tumour cells. Expansive foci of stromal haemorrhage, fibrosis and hemosiderin pigment deposits are observed5, 6. Elliptical tumour cells are bland with moderate, pale or faintly eosinophilic cytoplasm, an indistinct cellular perimeter and several intracytoplasmic, para-nuclear vacuoles. Nuclei are uniform, elliptical or stellate and imbued with fine nuclear chromatin with indistinct nucleoli. Pleomorphic, bizarre, mildly hyperchromatic nuclei with prominent nucleoli may be observed. Tumour necrosis is absent5, 6.
Angiomatoid fibrous histiocytoma exemplifies a diverse morphologic spectrum. The neoplasm consistently demonstrates sheets and miniature fascicles of elliptical, epithelioid or spindle-shaped cells with bland, vesicular nuclei. Tumour cells display a fibroblastic or histicytoid appearance and are incorporated with moderate, eosinophilic cytoplasm. Mitotic figures are infrequent and appear around 2 mitosis per 10 high power fields although atypical mitosis are absent 5, 6.
Lesions may demonstrate prominent, diffuse cellular and nuclear pleomorphism. However, cellular atypia and elevated mitotic figures are not associated with an inferior clinical outcome. Focal, intra-lesional haemorrhage configures blood-filled, pseudo-angiomatous vascular spaces of variable dimension. Occasionally, haemorrhagic vascular spaces configure a significant segment of tumefaction. Vascular spaces are devoid of an endothelial layer and are coated with flattened neoplastic cells. Hemosiderin pigment deposits can be prominent5, 6.
An estimated one third (33%) of neoplasms are solid and devoid of pseudo-angiomatous spaces. Enveloping stroma can be myxoid, sclerotic or desmoplastic-like, simulating a poorly differentiated carcinoma. Tumour giant cells and reactive, osteoclast-like giant cells can occasionally appear within pseudo-angiomatous spaces5, 6.
The multinodular neoplasm is composed of myoid, spindle-shaped or histiocytoid cells and configures a distinctive cellular syncytium. Pseudo- angiomatous, blood- filled vascular spaces are circumscribed by tumour cells along with an enveloping, thick, fibrous, pseudo-capsule. Tumefaction is encompassed with lymphocytic and plasma cell infiltrate with occasional articulation of germinal centres6.
Circumscribing, peri-capsular lymphoid infiltrate comprised of admixed B lymphocytes and T lymphocytes appears intermingled with scattered tumour cells, immune reactive to desmin4, 6
Despite diverse morphological configurations, the neoplasm may be devoid of characteristic, defining histological manifestations. Notwithstanding, a multinodular tumefaction composed of myoid, spindle-shaped cells is a consistent and diagnostic feature4.
Certain morphological features as nuclear grooving, occurrence of clear cells, rhabdomyoblast-like cells and clusters of miniature cells with minimal cytoplasm simulating cells of Ewing’s sarcoma or a reticular cell pattern entangled within a myxoid stroma are uncommon manifestations4, 5.
Schwannoma- like morphological features with nuclear palisading and prominent, stromal hyalinised vessels may be discerned. The neoplasm lacks a singular, confirmatory histological parameter which can appropriately surmise the biological behaviour of morphologically typical, primary or metastatic angiomatoid fibrous histiocytoma5, 6.
Hyperchromatic nuclei, significant nuclear atypia or enhanced mitotic activity is occasional and unrelated to an aggressive clinical course. However, angiomatoid fibrous histiocytoma may be associated with aberrant clinical and morphological features such as initial disease representation in the elderly, incrimination of extra-somatic soft tissues and modifications of tumour architecture, stromal matrix or cytological component. Myxoid variant of angiomatoid fibrous histiocytoma exhibits a predominant reticular configuration with prominent myxoid stroma6.
The variant exhibits morphological features such as a dense, fibrous pseudo-capsule, peripheral infiltrate of lymphocytes and plasma cells along with haemorrhage within cystic spaces5, 6.
Ultrastructural evaluation is devoid of pertinent cellular features. However, modified endothelial cells, smooth muscle cells, striated muscle cells, histiocyte- like cells, fibroblast- like cells and miniature, interdigitating cellular processes conjoined by desmosome- like junctions are discerned5, 6. Figure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7, Figure 8.
Figure 1.Angiomatoid fibrous histiocytoma demonstrating a multinodular pattern with circumscribing lymphoid and plasma cell infiltrate and a superimposed stratified squamous epithelium 9
Figure 2.Angiomatoid fibrous histiocytoma delineating sheets of spindle-shaped cells with bland nuclei and moderate cytoplasm disseminated in a vascularized stroma and a cuff of lymphoid cells 9.
Figure 3.Angiomatoid fibrous histiocytoma enunciating elliptical and spindle-shaped cells with moderate eosinophilic cytoplasm and intermixed lymphocytes and plasma cells 10.
Figure 4.Angiomatoid fibrous histiocytoma exhibiting spherical to elliptical cells comingled with abundant vascularized stroma and a peripheral accumulation of small lymphocytes with plasma cells 11.
Figure 5.Angiomatoid fibrous histiocytoma enunciating a multinodular pattern with elongated cells, vascular tumour stroma and a border of lymphocytes and plasma cells12.
Figure 6.Angiomatoid fibrous histiocytoma exemplifying uniform, spindle-shaped tumour cells with peripheral lymphocytic cuffing and a vascularized tumour matrix13.
Figure 7.Angiomatoid fibrous histiocytoma exhibiting spindle-shaped tumour cells with abundant eosinophilic cytoplasm, bland nuclei and a vascular tumour matrix 14.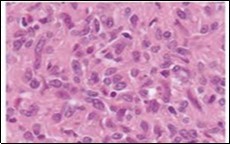
Figure 8.Angiomatoid fibrous histiocytoma depicting tumour nodules of elliptical cells with a cuff of lymphoid and plasma intermixed with a vascular matrix 15.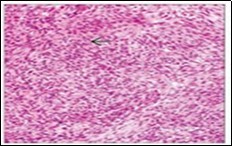
Immune Histochemical Elucidation
Angiomatoid fibrous histiocytoma lacks a pertinent panel of diagnostic immunohistochemistry and immune reactions are essentially non specific. An estimated 50% instances are diffusely or focally immune reactive to desmin and variably immune reactive to epithelial membrane antigen (EMA), CD99 and CD68. The neoplasm is infrequently immune reactive to myoid markers such as smooth muscle actin (SMA), calponin, h-caldesmon and CD217, 8.
Dendritic-like processes arising from tumour cells are immune reactive to desmin. Ki-67 labelling index is generally below < 5%. A subset of neoplasms delineate EWSR1 genetic rearrangements7, 8.
The neoplasm is immune non reactive to cytokeratin AE1/AE3, 8/18, myogenin, Myo-D1, melan-A, human melanoma black -45 (HMB-45) antigen, S100 protein, lysozyme, CD31,CD34, CD35, erythroblast transformation- specific (ETS)- related gene (ERG), glial fibrillary acidic protein (GFAP), factor VIII related antigen and p637, 8.
Differential Diagnosis
Angiomatoid fibrous histiocytoma necessitates differentiation from diverse reactive granulomas arising as a component of benign or malignant neoplasms. Specifically, sheets of elliptical or spindle-shaped cells may be consistently discerned with an absence of characteristic features such as a fibrous pseudo-capsule or lymphocytic and plasma cell infiltrate 2. Segregation is required from organized haematoma which appears as a misinterpretation of long standing neoplasms with extensive fibrosis, haemorrhage and deposition of hemosiderin pigment, thereby obscuring accumulation of neoplastic cells2, 4.
Granulomatous inflammation is usually devoid of vascular spaces and is composed of loosely cohesive, minimally solid zones of epithelioid cell aggregates 2, 4. aneurysmal benign fibrous histiocytoma is a superficial, dermal neoplasm occurring in young adults. The heterogeneous tumefaction enunciates typical architectural features of dermatofibroma, demonstrates giant cells and hemosiderin laden macrophages along with peripheral collagen bundles and hyperplasia of superimposed stratified squamous epithelium. A peri-capsular lymphocytic and plasma cell infiltrate is absent and the neoplasm is immune non reactive to desmin2, 4.
Spindle cell haemangioma is often localized within the dermis and subcutaneous tissue of extremities. The poorly circumscribed neoplasm demonstrates cavernous vascular articulations which are true vascular spaces layered by attenuated endothelial cells. A component of epithelioid cells and cells with intracytoplasmic vacuoles may be observed 2, 4.
Nodular Kaposi’s sarcoma is a well circumscribed, dermal neoplasm comprised of bland tumour cells and slit-like, blood- filled vascular configurations. Cystic, vascular zones are usually absent. Kaposi’s sarcoma is immune reactive to CD34 and human herpes virus 8 (HHV8)2, 4.
Palisaded myofibroblastoma occurring within lymph node is constituted by delicate palisades of spindle-shaped cells and dense bands of collagen fibres, designated as “amanthiod” fibres. The neoplasm is immune non reactive to desmin2, 4. inflammatory pseudo-tumour of lymph node is devoid of pseudo-vascular spaces 2, 4.
Follicular dendritic cell sarcoma usually arises within the lymph node, is immune reactive to CD21 or CD35 and is immune non reactive to desmin or smooth muscle actin (SMA) 2, 4.
Ewing’s sarcoma is morphologically diverse from angiomatoid fibrous histiocytoma wherein atypical Ewing’s sarcoma is composed of enlarged cells demonstrating cellular and nuclear pleomorphism. No age of disease emergence is exempt. Ewing’s sarcoma harbours chromosomal rearrangements of EWSR1-FLI1 and EWSR1 gene besides delineating adjunctive, typical genomic fusions 2, 4.
Rhabdomyosarcoma is a deep—seated, infiltrative neoplasm which can histologically recapitulate angiomatoid fibrous histiocytoma although it is devoid of a peripheral, capsular cuff of lymphocytes and plasma cells. The neoplasm exemplifies characteristic features of rhabdomyosarcoma with occurrence of rhabdomyoblasts or strap cells. Concomitant foci of embryonal or alveolar variant of rhabdomyosarcoma are discerned. The neoplasm is immune reactive to markers of skeletal muscle differentiation such as desmin, myogenin or MyoD12, 4.
Malignant extra-renal rhabdoid tumour is a frequently discerned, paediatric neoplasm which arises in children below < 3 years. The poorly circumscribed, infiltrative tumefaction is composed of sheets of enlarged, polygonal cells with eccentric, vesicular nuclei and intracytoplasmic, eosinophilic inclusions composed of aggregated intermediate filaments. Malignant extra-renal rhabdoid tumour is immune reactive to cytokeratin in a dot- like pattern and displays a loss of integrase interactor 1(INI1)2, 4.
Pleomorphic angiomatoid fibrous histiocytoma morphologically recapitulates an undifferentiated pleomorphic sarcoma, Nevertheless, undifferentiated sarcoma frequently arises within deep-seated soft tissue of elderly population and lacks characteristic genetic fusions discerned in classic angiomatoid fibrous histiocytoma2, 4.
Metastatic tumour deposits within a lymph node exhibits cytological atypia. Tumefaction may simulate angiomatoid fibrous histiocytoma on account of prominent circumscription by lymphoid cells. Also, metastatic deposits are encompassed by true, specific lymph node architecture with articulation of sub-capsular and medullary sinuses. Metastatic deposits are accompanied by history or occurrence of concurrent primary carcinoma. The condition can be discerned with appropriate clinical and radiological correlation. Primary angiomatoid fibrous histiocytoma within pulmonary parenchyma or brain tissue may be misinterpreted as metastatic deposits2, 4.
Primary pulmonary myxoid sarcoma requires segregation from endobronchial angiomatoid fibrous histiocytoma with a prominent reticular pattern. Primary pulmonary myxoid sarcoma is accompanied by a patchy, chronic lymphocytic and plasma cell infiltrate. Nevertheless, a predominant reticular tumour architecture and peripheral, capsular circumscription by lymphocytes and plasma cells is absent. The neoplasm is immune non reactive to desmin. Insignificant foci of myxoid stroma are encountered2, 4.
Myoepithelial tumour of soft tissue exhibits a reticular pattern and is associated with heterogeneous immune and phenotypic reactivity. Neoplasms are commonly discerned within second to fourth decade. Around one fifth (20%) of neoplasms arise in children. Commonly incriminated sites are extremities, shoulder girdle, trunk or head and neck. Myoepithelial tumours denominate diverse morphological patterns especially foci of ductular or tubular differentiation and chondromyxoid stroma 2, 4.
Myoepithelial neoplasia are immune reactive to epithelial membrane antigen (EMA), S100 protein and cytokeratin. Variable expression of glial fibrillary acidic protein(GFAP), smooth muscle actin (SMA), calponin and CD10 is observed. Roughly 50% of myoepithelial neoplasms depict EWSR1 genetic rearrangements with consequent genomic fusion of minimal partner genes such as POU5F1, PBX1 and ZNF44 gene. Additionally, association of soft tissue myoepithelial tumours with EWSR1-CREB1, EWSR1-ATF1 or FUS-ATF1 genetic fusion is undocumented.
Myoepithelial neoplasms depicting tubulo-ductal differentiation delineate reoccurring chromosomal rearrangements of PLAG1 as also described in pleomorphic adenoma of salivary gland. Myxoid angiomatoid fibrous histiocytoma requires segregation from diverse soft tissue tumours demonstrating a prominent myxoid matrix such as myoepithelioma, extra- skeletal myxoid chondrosarcoma and myxoid liposarcoma2, 3.
Myoepithelioma typically arises within the extremities. No age of tumour emergence is exempt. The multinodular tumefaction demonstrates diverse morphological configurations, a variable architecture, occurrence of epithelioid, elliptical or spindle-shaped cells, an encompassing chondromyxoid stroma and foci of cellular pleomorphism. Around 50% instances are immune reactive to epithelial membrane antigen (EMA). The neoplasm is immune reactive to myoepithelium- associated immune markers such as S100 protein and SOX10. Immune reactivity to desmin is exceptional2, 3.
Extra-skeletal myxoid chondrosarcoma is typically devoid of cartilaginous differentiation. The multinodular neoplasm depicts a fibrous pseudo-capsule, prominent myxoid matrix and significant stromal haemorrhage. Extra-skeletal myxoid chondrosarcoma appears in elderly population and exhibits a male predominance. Peri-capsular tumour circumscription with lymphocytes and plasma cells is absent. The neoplasm is immune non reactive to epithelial membrane antigen (EMA) and desmin. NR4A3 genetic rearrangement is commonly discerned2, 3.
Myxoid liposarcoma commonly arises within extremities of young adults. The multinodular neoplasm demonstrates a prominent myxoid matrix, arborizing “chicken- wire” vascular articulations, mucinous aggregates and foci of variable transition from myxoid to hyper-cellular zones. Myxoid liposarcoma lacks a fibrous pseudo-capsule, peripheral lymphocytic and plasma cell cuff or haemorrhage cystic spaces(2,3]. Lipoblasts are commonly enunciated. Myxoid liposarcoma is immune non reactive to desmin or epithelial membrane antigen (EMA). However, the neoplasm harbours genomic rearrangements of FUS and/or EWSR1 genes[3].
Low grade fibromyxoid sarcoma is a moderately cellular neoplasm composed of bland, fusiform or spindle-shaped cells with a focal whirling pattern, circumscription by dense, collagenous stroma with zonal transformation into myxoid areas. Foci of epithelioid cells, collagen rosettes and tumour cell infiltration into abutting skeletal muscle is observed. The neoplasm is immune reactive to MUC4, CD99, B cell lymphoma 2(bcl-2) antigen and epithelial membrane antigen (EMA)2, 3.
Investigative Assay
Fluorescent in situ hybridization (FISH) of tumour cells depicts a singular interphase nucleus in beyond > 10% of tumour cell nuclei, indicating an EWSR1 genetic rearrangement of chromosome 22. Fluorescent in situ hybridization (FISH) and reverse transcriptase polymerase chain reaction (RT-PCR) may be cogently employed to evaluate genetic configuration of angiomatoid fibrous histicytoma7, 8.
Therapeutic Options
The neoplasm can be appropriately managed with comprehensive surgical excision along with removal of a broad perimeter of uninvolved, tumour- free tissue. Aforesaid surgical manoeuvre is accompanied by a favourable prognosis. Adjuvant radiation therapy or chemotherapy is indicated in instances with distant metastasis or tumours unamenable to surgical resection, a requirement which can be evaluated with extensive tumour monitoring7, 8.
References
- 1.Enzinger F M. (1979) Angiomatoid malignant fibrous histiocytoma- a distinct fibrohistiocytic tumour of children and young adults simulating a vascular neoplasm” Cancer. 44, 2147-2157.
- 2.Thway K, Fisher C. (2015) Angiomatoid fibrous histiocytoma – the current status of pathology and genetics” Archives of pathology and laboratory medicine. 139(5), 674-682.
- 3.Ying L X, Teng X D. (2018) Myxoid and reticular anngiomatoid fibrous histiocytoma- a case confirmed by fluorescence in situ hybridization analysis for EWSR1 rearrangement”. , Int J Clin Exp Pathol 11(6), 3186-3190.
- 4.Saito K, Kobayashi. (2017) E et al” Angiomatoid fibrous histiocytoma – a series of seven cases including genetically confirmed aggressive cases and a literature review” BMC Musculoskeletal Disorders. 18-31.
- 5.Bohman S L, Goldblum J R. (2014) Angiomatoid fibrous histiocytoma – an expansion of the clinical and histological spectrum”. , Pathology 46, 199-204.
- 6.Thway K, Stefanaki K. (2013) Metastatic angiomatoid fibrous histiocytoma of the scalp with EWSR1-CREB1 gene fusion in primary tumour and nodal metastasis” Hum Pathol. 44(2), 289-293.
- 7.Schaefer I M, Fletcher C D. (2014) Myxoid variant of so-called angiomatoid “malignant fibrous histiocytoma clinicopathologic characterization in a series of 21 cases”. , Am J Surg Pathol 38, 816-823.